A causa della natura molto “volatile” dei genomi virali (e batterici), è molto difficile, se non impossibile, individuare i geni specifici che compongono una specie. I genomi umani, sebbene incredibilmente diversi nelle apparenze esteriori, sono super conservati rispetto ai genomi microbici.
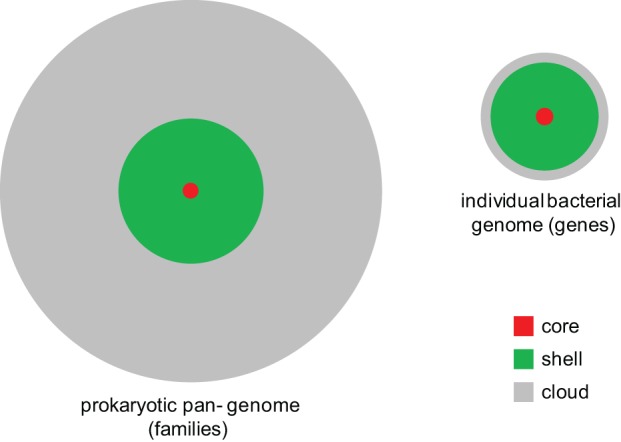
Come tale, genomica batterica e virale relativamente recente hanno avuto un piccolo cambiamento di paradigma. Invece di cercare di descrivere rigidamente un ceppo o una specie, lo definiscono come una nuvola di geni . Alcuni di questi geni sono essenziali per la specie / ceppo, quelli sono chiamati geni centrali. Intorno ai geni centrali, c'è una nuvola di geni opzionali. Ogni ceppo sceglie un gruppo di geni opzionali dalla nuvola, che verranno poi chiamati geni shell di quel ceppo specifico.
Ora sembra che una varietà sia un insieme molto specifico di geni shell + i geni principali. Ma in realtà, un ceppo stesso sarà una sorta di mini nuvola all'interno della nuvola con lievi variazioni nei geni meno importanti. Le differenze tra i ceppi sono quindi definite dalle variazioni nei geni più importanti. Questa non è tanto una regola rigida e basata sulla natura, ma più di “ciò che è importante per gli esseri umani”. Ad esempio, per noi esseri umani, la resistenza agli antibiotici è molto importante e quindi è probabile che definisca un ceppo. D'altra parte, una variazione del 46 esimo amminoacido di un enzima nella scomposizione dello zucchero che non influisce sull'efficienza non è molto importante e 'parte della nuvola.'


{kind=link}